Генетика остеопетроза
В январской онлайн-версии журнала "Current Osteoporosis Reports ( (2018) 16:13–25) опубликована обзорная статья итальянских специалистов, посвященная генетике остеопетроза.
Элеонора Палагано1,2, Сиро Менале1,3, Кристина Собакки1,3, Анна Вилла1,3
1Исследовательский клинический институт в Россано, Италия
2Отделение медицинских биотехнологий и трансляционной медицины, Миланский государственный университет, Милан, Италия
3 Национальный научно-исследовательский центр, Институт генетических и биомедицинских исследований, Милан, Италия
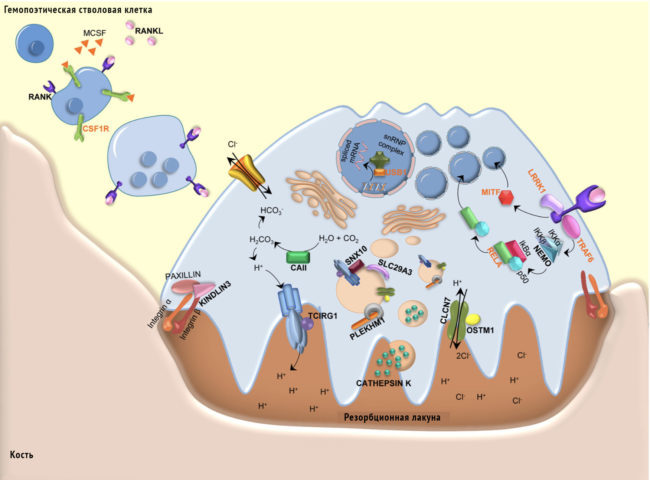
Схема молекул, участвующих в дифференциации и активации остеокластов и играющих роль в патогенезе остеопетроза. Наиболее известные гены заболевания выделены жирным шрифтом черного цвета. Недавно выявленные гены болезни, упомянутые в этом обзоре, выделены жирным шрифтом оранжевого цвета.
Цель обзора:
Термин «остеопетроз» объединяет в себе группу редких заболеваний скелета, характеризующихся одним общим признаком – увеличением плотности костной ткани вследствие нарушения ее резорбции. Остеопетроз – клинически и генетически гетерогенное заболевание, и в его прогнозировании и лечении немаловажную роль играет точная молекулярная классификация. В этой статье рассматриваются новые данные о патогенезе остеопетроза.
Новые данные:
Недавно опубликованные данные о новых мутациях известных генов, а также дефектах новых генов, расширяют наше представление о спектре молекулярных дефектов, приводящих к остеопетрозу.
Краткая справка:
Применение технологий секвенирования нового поколения постоянно расширяется, что способствует дифференциальной диагностике. Некоторые сложные фенотипы, при которых остеопетроз сопровождается дополнительными клиническими признаками, получили молекулярную классификацию, включая также новые гены. Кроме того, были распознаны новые типы мутаций, которые, по причине своего происхождения или расположения в геноме, было трудно обнаружить. Тем не менее, для некоторых паттернов мутации-провокаторы все еще остаются неизвестными, а значит имеется необходимость в дальнейшем активном изучении генетики остеопетроза.
Введение
Термин «остеопетроз» включает в себя группу редких наследственных заболеваний скелета, которые характеризуются заметным увеличением плотности костной ткани вследствие нарушения ее резорбции остеокластами – клетками, специально предназначенными для этой функции. Остеопетрозу присваивается одна из 3-ех форм на основании типа наследования: аутосомно-рецессивного (АРО), аутосомно-доминантного (АДО) и Х-сцепленного.
Заболеваемость AРО составляет 1 на 250 000 новорожденных, но в некоторых регионах (например, в Коста-Рике, Среднем Востоке, Чувашской республике РФ и Графстве Вестерботтон, Швеция) эта цифра выше из-за т.н. «эффекта основателя», географической изоляции или близкого родства родителей.
Наряду с термином «аутосомно-рецессивный остеопетроз» также используется термин «злокачественный ювенильный остеопетроз» (ЮЗО), т.к. данное заболевание диагностируется вскоре после рождения, и без лечения зачастую приводит к летальному исходу.
Заболеваемость АДО составляет 1 на 20 000 новорожденных. Его также называют поздней формой остеопетроза, поскольку болезнь впервые клинически проявляется в юности или во взрослом возрасте. АДО, в целом, считается доброкачественным заболеванием, т.к. продолжительность жизни пациентов обычно находится в пределах нормы. Однако степени тяжести АДО широко варьируются: от бессимптомного течения до случаев тяжелых поражений, если заболевание проявилось в раннем возрасте.
И, наконец, Х-сцепленный остеопетроз является крайне редкой формой, и за всю историю в литературе встречается всего несколько случаев среди неродственных пациентов.
Дефицит карбонгидразы II (CA II) – первая форма остеопетроза с установленным молекулярным патогенезом. Первоначальные свидетельства были получены по результатам биомедицинской оценки энзимной активности у пациентов. Затем с помощью прямого секвенирования гена CA II удалось определить конкретную мутацию. С 2000 года генетическая база остеопетроза значительно расширилась, что позволяет дать генетическую классификацию примерно в 90 % случаях. При этом некоторые случаи все еще нуждаются в точной диагностике. В случае «чистого» АРО, болезнь вызывают биаллельные мутации в одном из 7 различных генов: 5 из них (TCIRG1, CLCN7, OSTM1, SNX10, and PLEKHM1) кодируют протеины, задействованные в окислении резорбционных лакун и/или везикулярном транспорте. Мутации потери функции в данных генах приводят к остеопетрозу с избытком остеокластов, при котором остеокластов много, но они не функциональны. В то же время, мутации гена TNFSF11 (RANK) и его рецептора ассоциируются с АРО с дефицитом остеокластов, при котором остеокластогенез блокируется.
АДО I и II типа отличаются по ключевым клиническим признакам (основным очагам пониженной плотности костной ткани и предрасположенности к патологическим переломам) и генетическому дефекту, локализованному в генах LPR5 и CLCN7, соответственно. Тем не менее, поскольку АДО I типа развивается из-за нарушения активности остеобластов по причине снижения аффинности LPR5 к внеклеточным антагонистам SOST и DKK1 и последующей активации канонической сигнальной системы Wnt, было бы точнее классифицировать его как форму увеличения костной массы. По этой причине, последние литературные данные относительно мутаций гена LRP5 в данной работе не рассматриваются.
Кроме того, считается, что Х-сцепленный остеопетроз вызывают гипоморфические мутации в гене NEMO (эссенциальный модулятор NF-kB).
В данной статье рассмотрены самые последние генетические данные, расширяющие спектр молекулярных дефектов, приводящих к остеопетрозу. Вкратце описаны новые мутации, обнаруженные в вышеупомянутых генах. При этом особое внимание уделено новым типам мутаций, которые в некоторых случаях осложняют выбор вариантов по общепринятым критериям в ходе генетических исследований. Также представлены новые гены, ассоциированные с остеопетрозом у одного или нескольких пациентов. Молекулы и сигнальные пути, упомянутые в статье, как играющие роль в патогенезе остеопетроза, представлены в таблице 1.
Мутации известных генов
TCIRG1
Ген TCIRG1 (иммунорегулятор Т-клеток 1) кодирует a3-субъединицу V0-домена АТФ-зависимого протонного насоса V-АТФазы. Больше всего данный ген экспрессируется в остеокластах и париетальных клетках желудка: в костной ткани активность V-АТФазы требуется для достижения низкого pH, необходимого для растворения неорганического матрикса разрушения органического матрикса кости кислыми протеазами; в желудке она определяет низкий pH, необходимый для абсорбции пищевого Ca2+. Таким образом, это объясняет дефект минерализации кости и ассоциацию остеопетроза и рахита (т.н. остеопетрорахит) в результате мутации TCIRG1.
Помимо своей функции протонного насоса, V0-комплекс участвует в направленном перемещении пузырьков, фактически взаимодействуя с микротрубочками и актиновым цитоскелетом, возможно, непосредственно с помощью a3-субъединицы, и это играет важнейшую роль в формировании фестончатого края.
Мутации гена TCIRG1 ассоциируются с 50 % всех случаев АРО и распространяются по всему гену, вызывая нарушения функции протонного насоса V-АТФазы, а также перемещения/слияния пузырьков в остеокластах.
На сегодняшний день описано более 120 различных мутаций гена TCIRG1, включая миссенс-мутации, стоп-мутации, малые инсерции/делеции, крупные геномные делеции и дефекты сплайсинга, что свидетельствует о высокой генетической неоднородности когорты пациентов с TCIRG1-дефектным АРO. Этот факт также был подтвержден недавними отчетами об отдельных пациентах и небольших группах, являющихся носителями новых TCIRG1-мутаций. Недавно авторами статьи были опубликованы интересные сведения по данному вопросу: в первом отчете описаны 4 различных нуклеотидных изменения в 15 интроне, расположенном на удалении примерно 150 нуклеотидов от ближайшего участка канонического сплайсинга. По этой причине они получили название «глубокие интронные мутации». Данные мутации нарушают процесс сплайсинга из-за активации криптических сайтов сплайсинга. При этом нормальный сплайсинг прекращается не полностью, что может объяснить более мягкий фенотип в гомозиготном состоянии. Полученные сведения подчеркивают необходимость тщательной оценки возможного влияния интронных изменений в известных болезнетворных генах.
В другой статье описаны сходные мутации в 12 экзоне TCIRG1, который формирует внутренний акцепторный сайт сплайсинга в 12 экзоне, вызывая аберрацию сплайсинга, сдвиг рамки и преждевременную терминацию, о чем свидетельствует виртуальный скриниг и минигенные технологии. Подобный дефект был обнаружен у пациентов с АРО, вызванным мутацией гена CLCN7 (см. ниже). Полученные результаты согласуются с последними литературными данными.
Таблица 1. Гены, участвующие в патогенезе остеопетроза и смежных остеосклеротических расстройствах.
a Данные получены в ходе исследований на животных моделях
bПроцентная доля согласно генетическим данным в исследуемой когорте: 420 пациентов с АРО
сТГСК применима только при отсутствии прогрессирующей нейродегенерации.
CLCN7
Ген CLCN7 (потенциал-зависимый хлоридный канал-7) кодирует универсально экспрессируемый, медленный потенциал – зависимый канал-антипортер 2Cl− /1H+, который находится в мембране поздних эндосом и лизосом. Этот ген служит посредником в обмене хлоридных ионов и протонов, осуществляя связь с V-АТФазой в процессе окисления резорбционных лакун и везикул с лизосомами. CLCN7 выступает в качестве димера: каждый мономер содержит путь ионной транслокации с консервативным ионотропным глутаматным остатком и конформационные перестройки вне ионных путей приводят к синхронному открытию обоих каналов.
Рецессивные мутации гена являются причиной около 17 % всех случаев АРО, в то время как доминантные – причиной большинства случаев АДО II типа.
За последние годы несколько новых мутаций CLCN7 было обнаружено у пациентов из различных регионов (Китай, Тайвань, Япония, Эквадор, Италия, Марокко). Эти мутации ассоциировались со всеми известными формами CLCN7-зависимого остеопетроза: рецессивной, доминантной и промежуточной. Большинство новых мутаций являлись причиной изменения аминокислот и были обнаружены у единичных пациентов или целых семей. Фактически, это обусловило некоторую неопределенности в их интерпретации. Поэтому поиск оптимального способа исследования влияния CLCN7- мутаций на функции белков является насущной необходимостью, чтобы различать редкие полиморфизмы и мутации и правильно составлять корелляцию генотип-фенотип. Кроме того, как упоминалось выше, был выявлен синонимичный вариант в 12 экзоне CLCN7, вызывающий частичную аберрацию сплайсинга с пропуском 12 экзона внутри рамки. Полученные сведения привели к появлению гипотезы о том, что некоторые синонимичные, но не явно патогенные изменения способны модулировать фенотип мутаций CLCN7. Для подтверждения этой теории может понадобиться анализ клинических и генетических данных крупной когорты пациентов с CLCN7-зависимым остеопетрозом.
SNX10
Ген SNX10 (сортирующий нексин 10) кодирует белок, относящийся к семейству SNX – цитоплазматических и мембраносвязанных белков, характеризующихся наличием фосфоинозитид-связывающего домена PX. Как правило, SNX-белки участвуют в сортировке белков и мембранном транспорте, устанавливая связи белок-белок и белок-липид. В частности, SNX10 взаимодействует с V-АТФазой и регулирует ее внутриклеточный транспорт; таким образом, АРО с дефицитом SNX10 возникает в результате нарушения транспорта V-АТФазы к фестончатому краю и последующей дисфункции остеокластов. Также в очень редких случаях SNX10 может предположительно играть роль в транспорте и секреции матриксной металлпротеазы 9 при деградации внешнеклеточного матрикса.
Мутации гена SNX10 служат причиной около 5 % всех случаев АРО, в том числе т.н. Вестерботтенского остеопетроза (по названию провинции в Швеции с высокой частотой возникновения данного заболевания). В частности установлено, что вариант SNX10 c.212+1G>T, вызывающий активацию криптического сайта сплайсинга в 4-ом интроне и аберрантного сплайсинга, является общей мутацией для данной когорты пациентов, с необыкновенно высокой частотой носителей (1:93) в общей популяции Вестерботтена. Примечательно, что генеалогические исследования и гаплоидный анализ свидетельствуют в пользу наследования данной мутации от общих предшественников в начале 19-го века.
На клеточном уровне, несмотря на полученные ранее противоречивые данные по поводу наличия или нехватки остеокластов при АРО с дефицитом SNX-10, Статтин с соавт. говорят не о дефекте дифференциации остеокластов в МКПК пациентов, а о нарушении формирования фестончатого края. В то же время, остается под вопросом предположение, что у человека инактивация SNX10 приводит к остеопетрозному рахиту (о чем свидетельствуют исследования на мышиной модели), поскольку лишь у нескольких пациентов присутствует данный специфический фенотип. В очень редких случаях индуцированные плюрипотентные стволовые клетки (ИПСК) перепрограммируются из кожных фибробластов (у пациента с мутацией c.212+1G>T). Данные ИПСК могут способствовать изучению не раскрытых до конца особенностей SNX10-зависимого остеопетроза.
OSTM1
Ген OSTM1 (остеопетроз-ассоциированный трансмембранный белок 1) кодирует трансмембранный белок I типа, локализованный, в основном, на эндосомах и лизосомах. Он обладает высоко гликолизированным N-концом, который стабилизирует CLCN7 и защищает его от деградации лизосом, а также играет важную роль в обмене 2Cl− /1H+. Трансмембранный домен задействован в ионном обмене и CLCN7-зависимом транспорте к лизосомам. Предполагается также, что OSTM1 действует в качестве убиквитин-лигазы E3 для гетеротримерного G-белка Gαi3 и усиливает канонический сигнальный путь WNT с помощью модуляции взаимодействя β-катенин/ Lef1.
Недавно были выявлены дополнительные цитозольные партнеры OSTM1 по связыванию, что позволило предположить, что OSTM1 может служить в качестве адапторной молекулы внутри цитозольного поддерживающего мультипротеинового комплекса.
Мутации гена OSTM1 являются причиной примерно 5 % всех случаев АРО и всегда вызывают крайне тяжелый фенотип с быстро прогрессирующей первичной нейродгенерацией. Практически все выявленные мутации данного гена являются дефектами усечения. В связи с этим, при усечении OSTM1 в скрытой форме наблюдается ингибирование формирования остеокластов in vitro путем снижения числа осей BLIMP1-NFATc1, что, возможно, приводит к другим патогенным механизмам в случае АРО с дефицитом OSTM1. Более того,благодаря специально разработанному количественному ПЦР-анализу, две различных гомозиготных микроделеции, перекрывающие ~ 110 и ~ 10 кб соответственно и поражающие N-терминальный участок гена OSTM1, обнаружены у 5 тяжело больных пациентов из двух неродственных семей арабского и индийского происхождения. Анализ последовательностей релевантного геномного участка выявил AluSx-опосредованную рекомбинацию и нереккурентную перестройку, сопровождающуюся негомологичным соединением концов, в соответствии с лежащим в основе молекулярным механизмом.
Крайне редко у пациентов описывается остеопетроз с ранней нейродегенерацией и аккумуляцией железа в специфических областях головного мозга, что является крайне необычным случаем. Полное экзомное секвенирование выявило новые c.783+5G>T мутации гена OSTM1, вызывающие пропуск 4-го экзона, а также мутации сдвига рамки c.446dup в гомозиготном состоянии гена MANEAL. Данный ген кодирует эндо-альфа-подобный белок маннозидазы, который предположительно локализован в аппарате Гольджи и потенциально включен в метаболизм гликопротина; и действительно, в моче и цереброспинальной жидкости пациентов было обнаружено повышенное содержание маннозных молекул тетрасахариды. До сих пор неясно, как это соотносится с аккумуляцией железа в головном мозге. Влияние мутации гена MANEAL на фенотип, ассоциированный с OSTM1, в целом, требует дальнейшего изучения.
PLEKHM1
Ген PLEKHM1 (плекстрин гомологичное доменсодержащее семейство М – с RUN доменом 1) кодирует цитосолический белок, задействованный в эндосомальных транспортных путях ввиду взаимодейтсвия с малыми ГТФазами RAB7 и ARL8. Помимо этого, PLEKHM1 участвует в слиянии аутофагосом и лизосом, необходимом для клиренса различных белковых агрегатов. В соответствии с этим, разрушение специфичных PLEKHM1-доменов или потеря PLEKHM1 нарушает секрецию и доставку везикул, а также формирование фестончатого края, что ухудшает резорбтивную функцию остеокластов.
PLEKHM1 – это крупный белок, который содержит различные функциональные домены: домен RUN, где локализованы мутации (NM_014798.2:c.296+1G>A), изначально определенные у двух сиблингов с АРО; 2 плекстрин-гомологичных (PH) домена, отделенных друг от друга LC3-взаимодействующей областью (LIR); рубикон-гомологичный (RH) домен и «цинковый палец» С1 в карбоксильном конце. Две различные предположительно доминантные мутации гена PLEKHM1 выявлены у двух неродственных пациентов: мутация c.2140C>T:p.Arg714Cys, неявно ассоциированная с остеопетрозом, обнаружена во втором PH-домене; и недавно определенная мутация c.3051_3052delCA, расположенная в RH-домене и предположительно ликвидирующая мотив цинкового пальца. RH-домен необходим для взаимодействия PLEKHM1 с RAB7; в подтверждение этого, исследования сверхэкспрессии в клетках HEK293T показали снижение взаимодействия мутировавших белков с RAB7, приводящее к аномальной внутриклеточной локализации и повышению уровня аутофагии. Эти сведения внесли важный вклад в понимание функции PLEKHM1 для биологии костных клеток, несмотря на то, что некоторые аспекты и, в частности, взаимосвязь с аутофагией, остаются спорными и нуждаются в дальнейшем изучении.
CAII
Ген САII кодирует цитоплазмический фермент, катализирующий получение H2CO3 с помощью CO2 и H2O; затем, H2CO3 диссоциирует на HCO3 и ионы H+. Полученный H+ вытесняется V-АТФазой, в то время как HCO3 поглощается HCO3 − and H+ - анионным обменником, расположенным в базолатеральной мембране, которая предотвращает алкалинизацию цитоплазмы и обеспечивает ионы Cl−, необходимые для CLCN7/OSTM1 2Cl− /H+ антипортера.
Помимо того, ген CAII в больших количествах экспрессируется в почках и головном мозге. Фактически, пациенты с дефицитом CAII демонстрируют остеопетроз, почечный тубулярный ацидоз (ПТА) и церебральную кальцификацию, и данная триада нарушений сама по себе формирует диагноз. Примечательно то, что проксимальный ПТА недавно был описан также у пациента с АДО II, который является носителем распространенной при АДО II p.Gly215Arg мутации и у которого не было других мутаций, ассоциированных с ПТА; в данной ситуации точный патогенный механизм до сих пор остается неясным.
В процессе стандартизации условий для генетического тестирования выявлено около 30 различных мутаций CAII: миссенс-мутации, стоп-мутации и мутации сайта сплайсинга (среди последних - NM_000067.2:c.232+1G>A или т.н. арабская мутация, из-за широкой распространенности среди пациентов арабского происхождения). Большинство пациентов с мутацией CAII имеют арабское происхождение, однако встречаются также европейцы, испанцы, афро-амерканцы, азиаты и, реже, китайцы Хань.
FERMT3
Ген FERMT3 (член семейства фермитина 3) кодирует киндлин-3, член семейства киндлина, который включает 3 различных белка фокального контакта, задействованных в активации интегрина. Это процесс, необходимый для клеточных адгезии (слипания), распластывания и миграции, организации внутриклеточного матрикса, а также выживания, пролиферации и дифференциации клеток. Киндлин-3 – внутриклеточный белок, связанный с актиновым цитоскелетом. Он взаимодействует с множеством классов интегринов и способствует их адгезивной функции и сигнализации изнутри наружу, которая необходима костной ткани для резорбтивной функции остеокластов. Таким образом, дефицит киндлина-3 вызывает тяжелую морфологическую альтерацию остеокластов и нарушает их способность прилипать к поверхности костей. Интересен тот факт, что мутации гена FERMT3 ответственны за редкое аутосомно-рецессивное заболевание под названием дефицит адгезии лейкоцитов (LAD-III), некоторые пациенты с LAD-III также страдают от тяжелой формы остеопетроза (10 из 23 опубликованных случая). Эти мутации в основной массе являются высоко разрушительными: мутации усечения, дефекты сплайсинга, сдвиг рамки; из всей когорты выявлены только 2 миссенс-мутации. К сожалению, поскольку число описанных в литературе случаев ограничено, в настоящее время невозможно провести корреляцию генотип/фенотип.
RANKL
Ген RANKL (лиганд рецептора-активатора ядерного фактора каппа-B) предназначен для фундаментальных остеокластогенных цитокинов, которые путем связывания с его рецептором RANK запускают активацию нисходящего сигнального каскада, вызывающего дифференциацию и активацию остеокластов.
Недавно был найден дополнительный рецептор молекулы RANKL, богатый лейцином повтор-содержащий G-белок-связанный рецептор 4 (LGR4), который в результате связывания лигандов активирует сигнальный путь GSK3, тем самым подавляя экспрессию NFATc1 в ходе остеокластогенеза. Кроме того, были открыты ранее неизвестные и неожиданные функции RANKL в костной ткани, играющие важную роль в остеогенетической дифференциации мезенхимальных стволовых клеток, возможно, с помощью аутокринной петли.
Таким образом, полученные сведения представляют большой интерес для изучения АРО с дефицитом RANKL - редкой формы остеопетроза, которая составляет всего 2 % всех случаев. Для лучшего понимания упомянутых молекулярных механизмов и их значения в патофизиологии кости необходимы дальнейшие исследования.
По имеющимся данным, новых мутаций гена RANKL и соответственно случаев заболевания, помимо уже известных, до сих пор не было выявлено.
RANK
Как уже упоминалось выше, ген RANK (рецептор-активатор ядерного фактора каппа-B) является функциональным рецептором для RANKL.Связывание с лигандом вызывает тримеризацию рецептора и рекрутинг различных адапторных молекул, а также активацию различных сигнальных путей, таких как c-Jun N-терминальной киназы (JNK)/белок-активатор-1 (AP-1), ядерный фактор каппа-B (NF-kB) и ядерный фактор активированных Т-клеток с1 (NF-kB), Src и p38/MITF, Src and ERK, в результате чего происходит дифференциация, активация и выживание остеокластов. Мутации гена RANK ответственны приблизительно за 5% случаев АРО. В отличие от АРО с дефицитом RANKL, при котором костные дефекты не лечатся ТГСК, при АРО с дефицитом RANK ТГСК является эффективной, как показали недавно полученные данные.
SLC29A3
Дисостеосклероз – это редкая форма остеопетроза, которая развивается в младенчестве и проявляется типичными скелетными признаками: (расширение концов трубчатых костей и платиспондилия), а также различными поражениями кожи. Клиническое течение и прогнозы, как правило, довольно благоприятные. Имеющиеся литературные данные касательно генетики диостеосклероза довольно ограничены. Ген SLC29A3 (глюкозный траспортер-3 семейства 29), который при дисостеосклерозе поражен, кодирует нуклеотидный транспортер лизосом, в больших количествах вырабатываемый в клетках миелоидной линии. Описываемые мутации (NM_018344:c. 607T>C:p.Ser203Pro и c.1157G>A:p.Arg386Gln у одного пациента и c.1346C>G:p.Thr449Arg у второго в гомозиготном состоянии, а также c.303_320dup:p.102_107dup в гомозиготном состоянии) могут оказывать влияние на функцию и дифференциацию остеокластов, на что указывает сниженное число остеокластов после дифференциации in vitro МКПК пациентов, а также образцов биопсии кости. Мутации SLC29A3 также ассоциируются с гистиоцитозом с массивной лимфаденопатией – группой заболеваний, не затрагивающих или лишь слегка затрагивающих скелет. Таким образом, необходимы дальнейшие исследования для лучшего понимания роли данного гена в костной ткани, и особенно – в патофизиологии в целом.
CTSK
Пикнодизостоз - это редкая аутосомно-рецессивная скелетная дисплазия (примерная частота случаев - 1:1,7 млн. человек), которую включают в дифференциальную диагностику остеопетроза из-за повышенной плотности костной ткани в трубчатых костях. Другие типичные для пикнодизостоза клинические признаки включают низкорослость, которую с переменным успехом можно лечить гормонами роста, незакрытые родничок и швы черепа, переломы, тупой нижнечелюстной угол и акроостеолиз дистальной фаланги. При данном заболевании поражен ген CTSK, кодирующий катепсин К – цистеиновую пептидазу суперсемейства папаин, используемую остеокластами для деградации костного матрикса и обладающую уникальной способностью расщеплять молекулы коллагена на маленькие фрагменты. Кроме того, недавно было обнаружено, что катепсин К расщепляет и активирует матриксную металлопротеиназу 9 in vitro, что может указывать на наличие протеаз-сигнальной сети, вероятно релевантной для ряда физиопатологических нарушений. Также последние исследования показали, что катепсин-K играет роль в регуляции моделирования кости путем деградации периостина - белка внутриклеточного матрикса кортикального слоя, необходимого для формирования надкостницы при участии сигнального пути Wnt-β-катенина.
До сих пор в литературе было описано около 60 различных мутаций CTSK у пациентов из различных географических регионов. Наиболее частыми вариантами являются миссенс-мутации, также выявлены сдвиг рамки, нонсенс-мутации и дефекты сплайсинга. Мутации возникают, в основном, в зрелых CTSK-белках, с «горячими точками» в 5 и 6 экзонах.
Кроме того, около 6% мутаций обнаружено в пререгионе и 25 % - в прорегионе – небольших N-терминальных доменах, необходимых, соответственно, для правильной локализации и укладки белков и внутриклеточного транспорта. Прорегион также важен для поддержания ферментов в неактивном состоянии, при низком уровне pH он расщеплен. При этом корреляция генотип-фенотип до сих пор остается неизученной до конца.
Гены, вовлеченные в недавно открытые синдромные формы остеопетроза
TRAF6
Наряду с различными адапторными молекулами, необходимыми для связывания RANKL/ RANK, TRAF6 (TNF-рецептор-ассоциированный-фактор-6) представляется наиболее важной. Действие TRAF6 также снижает продукцию рецепторов Т- и B-клеток, что приводит к активации NF-kB. Несколько лет назад было определено, что деактивация гена TRAF6 у мышей вызывает тяжелый остеопетроз, и, крайне редко, схожее действие наблюдалось и у человека. Фактически, гомозиготная делеция 2064 Kb в 11-ой хромосоме, перекрывающая 5 регион в генах TRAF6, RAG1 и RAG2 (белки RAG необходимы для рекомбинации рецепторов B и T клеток, а также для их выживания и дифференциации) обнаружена у 2 сиблингов с остеопетрозом и тяжелым комбинированным иммунодефицитом (ТКИД) с помощью хромосомного микроматричного анализа. Точные границы делеции определены не были; что же касается TRAF6, его геномная делеция включала регион выше 1 экзона и часть некодирующей последовательности экзона 1. Данные участки с большой вероятностью являются регуляторными. Фактически, на белковом уровне их делеция полностью отменяет продукцию TRAF6. Результирующий фенотип является специфичным, поскольку остеопетроз у пациентов с данной группой мутаций был выражен исключительно в костях таза и ног. Поскольку в обоих случаях летальный исход наступил в очень раннем возрасте в связи с тяжелым иммунодефицитом и в настоящее время подобные случаи неизвестны, проследить развитие скелетного заболевания с данной генетической базой затруднительно.
LRRK1
Остеосклеротичная метафизарная дисплазия является формой остеопетроза, поражающей метафиз длинных костей, концевые пластинки позвонков, оконечности ребер, а также края плоских костей, в то время как череп остается непораженным. Описано только 5 случаев данного заболевания; в одном из них недавно была определена гомозиготная делеция семи нуклеотидов в последнем экзоне гена LRRK1 (лейцин-богатая повторная киназа 1) (NM_024652:c.5938_5944delGAGTGGT:p.Glu1980Alafs*66), предположительно вызвавшая сдвиг рамки и преждевременную терминацию, с потерей седьмой триптофан-аспартической кислоты (WD) 40-го домена. Домен WD-40, также как остальные функциональные домены белка LRRK1, выступает посредником во взаимодействии белок-белок. В частности, LRRK1 предположительно взаимодействует с компонентом сигнального пути c-Src для достижения цитоскелетной перестройки и формирования фестончатого края и подосомы. Таким образом, у мышей остеокласты с дефицитом Lrrk1 являются крупными и плоскими, поскольку они неспособны должным образом осуществить реорганизацию цитоскелета и резорбцию кости. Ген LRRK1, в целом, представляет высокий интерес для костной биологии и дальнейшие исследования должны определить его физиопатологическую роль.
MITF
Микрофтальмия-ассоциированный транскрипционный фактор (MITF) представляет собой транскрипционный фактор с основным доменом типа спираль-петля-спираль (лейциновая застежка). Он формирует гомо- и гетеродимеры, регулирующие генную экспрессию в различных тканях. По этой причине его мутация может вызвать широкий спектр фенотипов. В костной ткани MITF предположительно действует совместно с сигнальным путем RANKL/RANK, уменьшая продукцию NFATc1 для усиления NFATc1-зависимого остеокластогенного сигнала. И действительно, для мышей с Mitf mi/mi характерен остеопетроз в связи с дефектом остеокластогенеза на ранних стадиях.
Сложные гетерозиготные мутации гена MITF совсем недавно были обнаружены у двух неродственных пациентов с т.н. COMMAD-синдромом (колобома, остеопетроз, микрофтальмия, макроцефалия, альбинизм и глухота). Выявленные мутации (NM_198159.2:c.952_ 954delAGA:p.Arg318del and c.921G>C:p.Lys307Asn у пробанда I; c.952A>G:p.Arg318Gly и c.938- 1G>A:p.Leu312fs у пробанда II) повлияли не на димеризацию MITF, а скорее на его миграцию внутри ядра и ДНК-связывающие свойства. Данное открытие указывает на ряд фенотипов, связанных с MITF; фактически, в отличие от вариантов с рецессивными мутациями, доминантные ассоциированы с синдромом Ваандербурга типа 2A и синдромом Тица, для которых характерны глухота и пигментный дефицит. В целом, эти данные подтверждают ключевую роль MITF в процессах развития, а также дифференциации и выживания клеток.
Сигнальный путь NF-kB
Сигнальный путь NF-kB включает ряд молекул (в основном, киназы и транскрипционные факторы), которые играют ключевую роль в экспрессии регуляторных генов во многих органах и физиопатологических процессах. В костной ткани на это указывает тот факт, что гипоморфные мутации гена NEMO, кодирующего компонент IkB-киназного комплекса, необходимого для ингибирования IkB-α и последующей ядерной транслокации продуцируемого гетеродимера p65/p50, ответственны за Х-сцепленный остеопетроз с эктодермальной дисплазией и иммунодефицитом. Данные мутации в основном локализованы в белковом домене «цинковый палец» и приводят к остеопетрозу по причине альтерации сигнального пути RANKL/RANK.
Недавно было выявлено, что белок p65 (Rela) сам по себе связан с фенотипом, характеризующимся высокой костной массой. Фактически, в случае с внезапным летальным исходом новорожденного пациента, патологическое увеличение костной массы, выявленное в результате аутопсии и обоснованное повышенной функцией остеобластов, как оказалось, было ассоциировано с de novo миссенс-мутацией гена RELA (NM_021975.3:c.1534_ 1535delinsAG:p.Asp512Ser). Данная мутация, по наблюдениям, нарушает сигнализацию NF-kB в фиробластах пациентов, и возможно, может привести к нарушениям различных жизненно важных функций.
CSF1R
Наряду с RANKL, M-CSF является ключевой остеокластогенной молекулой. Это можно явно наблюдать при остеопетрозе с нехваткой остеокластов у мышей с дефицитом данного цитокина (мышиная модель op/op). У мышей дефицит M-CSF-рецептора (CSF1R) приводит к сходному остеопетрозному фенотипу; кроме того, обе модели демонстрируют дефекты врожденного иммунитета, фертильности и неврологической функции.
Примечательно то, что доминантные мутации гена CSF1R вызывают взрослую форму энцефаломиопатии, в то время как совсем недавно подозревалось, что именно рецессивная мутация данного гена ответственна за летальный, тяжелый фенотип у 2 сиблингов с генерализированным остеопетрозом и мальформацией мозга в тяжелой форме.
Вкратце, секвенирование экзома близкородственных родителей выявило гетерозиготную мутацию усечения (NM_001288705.1:c.1620C>T:p.Tyr540*) в гене CSF1R, который предположительно активировал белок с нехваткой внутриклеточного домена, необходимого для лиганд-зависимой димеризации и аутофосфореляции. В условиях отсутствия образцов ДНК, гомозиготность мутации CSF1R у пациентов не могла быть продемонстрирована; по этой причине, полученные результаты нельзя считать полностью обоснованными. Тем не менее, на их основании возможно проанализировать гены других пациентов со сходным фенотипом, чтобы попытаться определить дополнительные мутации в качестве подтверждения.
C16orf57
Несколько лет назад был описан случай итальянского пациента, у которого был обнаружен остеопетроз, ассоциированный с пойкилодермией и нейтропенией (ПН). ПН-синдром - это наследственный генодерматоз, который характеризуется ранней пойкилодермией, дистрофией ногтей, ладонно-подошвенным гиперкератозом и устойчивой нейтропенией, приводящей к рекуррентным инфекциям. В подобных случаях могут присутствовать скелетные дефекты, такие, как остеопения, краниофациальный дисморфизм, переломы, постнатальная задержка созревания и роста скелета. Однако случай, описанный Мильяччо с соавт. оказался нетипичным. ПН-синдром был обусловлен мутациями гена C16orf57, кодирующего фосфодиэстеразу, ответственную за модификацию и стабилизацию U6 небольшой ядерной РНК (USB1), которая является необходимым элементом структуры сплайсомы.
Позднее у пациента с остопетрозом и ПН была обнаружена гомозиготная стоп-мутация в гене C16orf57 (ENSG00000103005:c.232C>T:p.Arg78*), предположительно вызвавшая серьезные нарушения укладки процессированного белка. Выявленный патогенетический механизм может быть неполным или аберрантным сплайсингом специфических классов генов, как можно предполагать учитывая нейтропению. Тем не менее, для подтверждения данной гипотезы необходимы дальнейшие исследования скелетных клеток.
Современные методы терапии
В настоящее время единственным утвержденным методом лечения АРО является трансплантация гемопоэтических стволовых клеток (ТГСК). Остеокласты, фактически, являются клетками гемопоэтического происхождения, поэтому ТГСК способствует восстановлению костной резорбции с помощью донорских клеток. Данная статья не предусматривает подробного описания метода ТГСК: режимов кондиционирования, источников ГСК, типов донорства, графика и других, без сомнения важных, аспектов. Подробную информацию о них можно найти в недавно вышедших работах мировых экспертов. Здесь будут лишь вкратце приведены некоторые концепции.
Во-первых, молекулярные исследования вносят важный вклад в определение того, в каких случаях следует проводить ТГСК. Фактически, пациенты с мутациями RANKL или OSTM1 не могут быть кандидатами на данную процедуру: в первой группе дефекты не являются клеточно-автономными и поэтому не могут быть скорректированы с помощью ТГСК, а во второй у пациентов присутствовали серьезные неврологические нарушения несовместимые с жизнью. Для подобных случаев в настоящее время терапии, к сожаллению, не существует.
Кроме того, тяжесть клинической картины диктует метод лечения: легкие формы, ассоциированные с PLEKHM1, SLC29A3 или CTSK или доминантными мутациями CLCN7 не являются основанием для рискованных инвазивных процедур, проводимых при ТГСК. В таких случаях применяется только симптоматическое лечение в случае осложнений (переломов, дефектов зубов, отоларингологических проблем). С другой стороны, при АРО с дефицитом CLCN7 рекомендована тщательная неврологическая оценка с целью принятия решения о релевантности ТГСК. Установленная прогрессивная дегенерация препятствует проведению трансплантации. К сожалению, в настоящий момент, в данной субгруппе не установлена специфичная корелляция генотип-фенотип, позволяющая выявить пациентов, подходящих для ТГСК на основании лишь молекулярных данных.
Наконец, что касается недавно диагностированных форм заболевания, пока недостаточно данных для определения специфической стратегии лечения в дополнение к основной.
Выводы
Последние данные подтверждают генетическую неоднородность остеопетроза, осложняющую молекулярную диагностику. Фактически, в патогенез остеопетроза включены более 10 генов, включая самые крупные. Среди наиболее часто поражаемых определено большое количество различных мутаций: некоторые из них присутствуют у многих пациентов различного происхождения, другие - в единичных случаях, также определены новые мутации. По этим причинам, секвенирование следующего поколения (NGS), включая полное экзомное секвенирование (WES) и таргетированное секвенирование с использованием аналитических панелей, прочно вошло в клиническую практику в качестве стандартной процедуры диагностики остеопетроза. Комплексный метод имеет явные преимущества в скорости и более приближен к точной молекулярной диагностики, необходимой для прогнозирования и лечения, по сравнению с классическим секвенированием по Сенгеру генов-«кандидатов».
Кроме того, с помощью WES в единичных случаях у пациентов с нетипичными фенотипами удалось определить мутации в новых генах, которые, как уже известно играют роль в гомеостазе костной ткани: TRAF6, LRRK1, MITF, CSF1R и RELA. Генетические данные при этом указывают на возможную, но еще до конца не определенную роль C16orf57 в костях. В некоторых случаях, базисный патогенный механизм все еще нуждается в дальнейшем изучении, несмотря на то, что по литературным данным можно достаточно обоснованно судить об отрицательном воздействии выявленной мутации. С другой стороны, 2 класса мутаций, а именно глубокие интронные или синонимичные изменения, демонстрируют возможные ограничения WES. На самом деле, даже несмотря на то, что многие некодирующие участки являются целью WES, стандартный процесс по отбору вариантов рассматривает только интронные изменения, близкие к каноничным сайтам сплайсинга; в особенности, тенденцию оставаться незамеченными имеют варианты в конце прочтения.
Поэтому, если мутация локализована в данных регионах, она может быть пропущена. Фактически, данная работа ясно демонстрирует у 2 неродственных семей патогенный эффект глубоких интронных однонуклеотидных изменений, обнаруженных в гене TCIRG1, который сначала был пропущен WES. Схожим образом синонимичные изменения часто отфильтровываются в ходе стандартного технологического процесса при WES. На самом деле, они часто игнорируются также при стандартном секвенировании по Сенгеру, поскольку предположительно являются «тихими». Испытуемая группа продемонстрировала ( и это совпадает с результатами других исследований), что данное предположение в некоторых случаях может быть в корне неверным.
В заключении можно отметить, что совершено значительное достижение в изучении генетической базы остеопетроза. Тем не менее, около 10 % случаев все еще нуждаются в молекулярной классификации и у данных пациентов могут быть обнаружен мутации новых, ранее неизвестных генов либо неявные мутации в известных. Для того, чтобы восполнить пробелы, необходимо найти стратегию по интеграции NGS-технологий и улучшению способности интерпретировать геномные данные. В комплексе это позволит определить клинически релевантные варианты среди десятков тысяч в каждом отдельном экзоне.
Элеонора Палагано1,2, Сиро Менале1,3, Кристина Собакки1,3, Анна Вилла1,3
1Исследовательский клинический институт в Россано, Италия
2Отделение медицинских биотехнологий и трансляционной медицины, Миланский государственный университет, Милан, Италия
3 Национальный научно-исследовательский центр, Институт генетических и биомедицинских исследований, Милан, Италия
Схема молекул, участвующих в дифференциации и активации остеокластов и играющих роль в патогенезе остеопетроза. Наиболее известные гены заболевания выделены жирным шрифтом черного цвета. Недавно выявленные гены болезни, упомянутые в этом обзоре, выделены жирным шрифтом оранжевого цвета.
Цель обзора:
Термин «остеопетроз» объединяет в себе группу редких заболеваний скелета, характеризующихся одним общим признаком – увеличением плотности костной ткани вследствие нарушения ее резорбции. Остеопетроз – клинически и генетически гетерогенное заболевание, и в его прогнозировании и лечении немаловажную роль играет точная молекулярная классификация. В этой статье рассматриваются новые данные о патогенезе остеопетроза.
Новые данные:
Недавно опубликованные данные о новых мутациях известных генов, а также дефектах новых генов, расширяют наше представление о спектре молекулярных дефектов, приводящих к остеопетрозу.
Краткая справка:
Применение технологий секвенирования нового поколения постоянно расширяется, что способствует дифференциальной диагностике. Некоторые сложные фенотипы, при которых остеопетроз сопровождается дополнительными клиническими признаками, получили молекулярную классификацию, включая также новые гены. Кроме того, были распознаны новые типы мутаций, которые, по причине своего происхождения или расположения в геноме, было трудно обнаружить. Тем не менее, для некоторых паттернов мутации-провокаторы все еще остаются неизвестными, а значит имеется необходимость в дальнейшем активном изучении генетики остеопетроза.
Введение
Термин «остеопетроз» включает в себя группу редких наследственных заболеваний скелета, которые характеризуются заметным увеличением плотности костной ткани вследствие нарушения ее резорбции остеокластами – клетками, специально предназначенными для этой функции. Остеопетрозу присваивается одна из 3-ех форм на основании типа наследования: аутосомно-рецессивного (АРО), аутосомно-доминантного (АДО) и Х-сцепленного.
Заболеваемость AРО составляет 1 на 250 000 новорожденных, но в некоторых регионах (например, в Коста-Рике, Среднем Востоке, Чувашской республике РФ и Графстве Вестерботтон, Швеция) эта цифра выше из-за т.н. «эффекта основателя», географической изоляции или близкого родства родителей.
Наряду с термином «аутосомно-рецессивный остеопетроз» также используется термин «злокачественный ювенильный остеопетроз» (ЮЗО), т.к. данное заболевание диагностируется вскоре после рождения, и без лечения зачастую приводит к летальному исходу.
Заболеваемость АДО составляет 1 на 20 000 новорожденных. Его также называют поздней формой остеопетроза, поскольку болезнь впервые клинически проявляется в юности или во взрослом возрасте. АДО, в целом, считается доброкачественным заболеванием, т.к. продолжительность жизни пациентов обычно находится в пределах нормы. Однако степени тяжести АДО широко варьируются: от бессимптомного течения до случаев тяжелых поражений, если заболевание проявилось в раннем возрасте.
И, наконец, Х-сцепленный остеопетроз является крайне редкой формой, и за всю историю в литературе встречается всего несколько случаев среди неродственных пациентов.
Дефицит карбонгидразы II (CA II) – первая форма остеопетроза с установленным молекулярным патогенезом. Первоначальные свидетельства были получены по результатам биомедицинской оценки энзимной активности у пациентов. Затем с помощью прямого секвенирования гена CA II удалось определить конкретную мутацию. С 2000 года генетическая база остеопетроза значительно расширилась, что позволяет дать генетическую классификацию примерно в 90 % случаях. При этом некоторые случаи все еще нуждаются в точной диагностике. В случае «чистого» АРО, болезнь вызывают биаллельные мутации в одном из 7 различных генов: 5 из них (TCIRG1, CLCN7, OSTM1, SNX10, and PLEKHM1) кодируют протеины, задействованные в окислении резорбционных лакун и/или везикулярном транспорте. Мутации потери функции в данных генах приводят к остеопетрозу с избытком остеокластов, при котором остеокластов много, но они не функциональны. В то же время, мутации гена TNFSF11 (RANK) и его рецептора ассоциируются с АРО с дефицитом остеокластов, при котором остеокластогенез блокируется.
АДО I и II типа отличаются по ключевым клиническим признакам (основным очагам пониженной плотности костной ткани и предрасположенности к патологическим переломам) и генетическому дефекту, локализованному в генах LPR5 и CLCN7, соответственно. Тем не менее, поскольку АДО I типа развивается из-за нарушения активности остеобластов по причине снижения аффинности LPR5 к внеклеточным антагонистам SOST и DKK1 и последующей активации канонической сигнальной системы Wnt, было бы точнее классифицировать его как форму увеличения костной массы. По этой причине, последние литературные данные относительно мутаций гена LRP5 в данной работе не рассматриваются.
Кроме того, считается, что Х-сцепленный остеопетроз вызывают гипоморфические мутации в гене NEMO (эссенциальный модулятор NF-kB).
В данной статье рассмотрены самые последние генетические данные, расширяющие спектр молекулярных дефектов, приводящих к остеопетрозу. Вкратце описаны новые мутации, обнаруженные в вышеупомянутых генах. При этом особое внимание уделено новым типам мутаций, которые в некоторых случаях осложняют выбор вариантов по общепринятым критериям в ходе генетических исследований. Также представлены новые гены, ассоциированные с остеопетрозом у одного или нескольких пациентов. Молекулы и сигнальные пути, упомянутые в статье, как играющие роль в патогенезе остеопетроза, представлены в таблице 1.
Мутации известных генов
TCIRG1
Ген TCIRG1 (иммунорегулятор Т-клеток 1) кодирует a3-субъединицу V0-домена АТФ-зависимого протонного насоса V-АТФазы. Больше всего данный ген экспрессируется в остеокластах и париетальных клетках желудка: в костной ткани активность V-АТФазы требуется для достижения низкого pH, необходимого для растворения неорганического матрикса разрушения органического матрикса кости кислыми протеазами; в желудке она определяет низкий pH, необходимый для абсорбции пищевого Ca2+. Таким образом, это объясняет дефект минерализации кости и ассоциацию остеопетроза и рахита (т.н. остеопетрорахит) в результате мутации TCIRG1.
Помимо своей функции протонного насоса, V0-комплекс участвует в направленном перемещении пузырьков, фактически взаимодействуя с микротрубочками и актиновым цитоскелетом, возможно, непосредственно с помощью a3-субъединицы, и это играет важнейшую роль в формировании фестончатого края.
Мутации гена TCIRG1 ассоциируются с 50 % всех случаев АРО и распространяются по всему гену, вызывая нарушения функции протонного насоса V-АТФазы, а также перемещения/слияния пузырьков в остеокластах.
На сегодняшний день описано более 120 различных мутаций гена TCIRG1, включая миссенс-мутации, стоп-мутации, малые инсерции/делеции, крупные геномные делеции и дефекты сплайсинга, что свидетельствует о высокой генетической неоднородности когорты пациентов с TCIRG1-дефектным АРO. Этот факт также был подтвержден недавними отчетами об отдельных пациентах и небольших группах, являющихся носителями новых TCIRG1-мутаций. Недавно авторами статьи были опубликованы интересные сведения по данному вопросу: в первом отчете описаны 4 различных нуклеотидных изменения в 15 интроне, расположенном на удалении примерно 150 нуклеотидов от ближайшего участка канонического сплайсинга. По этой причине они получили название «глубокие интронные мутации». Данные мутации нарушают процесс сплайсинга из-за активации криптических сайтов сплайсинга. При этом нормальный сплайсинг прекращается не полностью, что может объяснить более мягкий фенотип в гомозиготном состоянии. Полученные сведения подчеркивают необходимость тщательной оценки возможного влияния интронных изменений в известных болезнетворных генах.
В другой статье описаны сходные мутации в 12 экзоне TCIRG1, который формирует внутренний акцепторный сайт сплайсинга в 12 экзоне, вызывая аберрацию сплайсинга, сдвиг рамки и преждевременную терминацию, о чем свидетельствует виртуальный скриниг и минигенные технологии. Подобный дефект был обнаружен у пациентов с АРО, вызванным мутацией гена CLCN7 (см. ниже). Полученные результаты согласуются с последними литературными данными.
Таблица 1. Гены, участвующие в патогенезе остеопетроза и смежных остеосклеротических расстройствах.
| Ген | Нарушаемая функция остеокластов | Частота | Тип остеопетроза | Терапия |
| TCIRG1 | Кислотная секреция Везикулярный транспорт | 50 %b | С избытком остеокластов | ТГСК |
| CLCN7 | Окисление лизосомального транспорта | 17,5b (APО) 80 % (АДО) | С избытком остеокластов | ТГСК для случаев АДОс, симптоматическое лечение |
| SNX10 | Эндолизосомальный транспорт/слияние | 4,5 %b | С избытком остеокластов | ТГСК |
| OSTM1 | Окисление лизосомального транспорта | 5 %b | С избытком остеокластов | В настоящее время не существует |
| PLEKHM1 | Эндолизосомальный транспорт/слияние | 2 пац.b (АРО) 2 пац. (АДО) | С избытком остеокластов | Симптоматическое лечение |
| CAII | Окисление | <1:106 | С избытком остеокластов | ТГСКс |
| FERMT3 | Слипание и распластывание | 10 пац. | С избытком остеокластов | ТГСК |
| RANKL | Дифференциация остеокластов | 2 %b | С дефицитом остеокластов | В настоящее время не существует |
| RANK | Дифференциация остеокластов | 4.5 %b | ТГСК | |
| SLC29A3 | Эндолизосомальная функция | 3 пац. | С дефицитом остеокластов | подлежит уточнению |
| TRAF6 | Слипание и резорбцияa | 2 пац. | С дефицитом остеокластов | подлежит уточнению |
| LRRK1 | Зона уплотнения и область формирования фестончатого краяa | 1 пац. | С избытком остеокластов | подлежит уточнению |
| MITF | Контроль генной экспрессииa | 2 пац | С избытком остеокластов | подлежит уточнению |
| NEMO | Oc-дифференциация/активация | 6 пац. | С избытком остеокластов | ТГСК |
| RELA | Ос-дифференциация | 1 пац. | С избытком остеокластов | подлежит уточнению |
| CSFIR | Ос-дифференциация | 2 пац. | С дефицитом остеокластов | подлежит уточнению |
| C16ORF57 | Подлежит уточнению | 2 пац. | С избытком остеокластов | Симптоматическое лечение |
| CTSK | Деградация костного матрикса | 1-1,7:106 | С избытком остеокластов | Симптоматическое лечение |
a Данные получены в ходе исследований на животных моделях
bПроцентная доля согласно генетическим данным в исследуемой когорте: 420 пациентов с АРО
сТГСК применима только при отсутствии прогрессирующей нейродегенерации.
CLCN7
Ген CLCN7 (потенциал-зависимый хлоридный канал-7) кодирует универсально экспрессируемый, медленный потенциал – зависимый канал-антипортер 2Cl− /1H+, который находится в мембране поздних эндосом и лизосом. Этот ген служит посредником в обмене хлоридных ионов и протонов, осуществляя связь с V-АТФазой в процессе окисления резорбционных лакун и везикул с лизосомами. CLCN7 выступает в качестве димера: каждый мономер содержит путь ионной транслокации с консервативным ионотропным глутаматным остатком и конформационные перестройки вне ионных путей приводят к синхронному открытию обоих каналов.
Рецессивные мутации гена являются причиной около 17 % всех случаев АРО, в то время как доминантные – причиной большинства случаев АДО II типа.
За последние годы несколько новых мутаций CLCN7 было обнаружено у пациентов из различных регионов (Китай, Тайвань, Япония, Эквадор, Италия, Марокко). Эти мутации ассоциировались со всеми известными формами CLCN7-зависимого остеопетроза: рецессивной, доминантной и промежуточной. Большинство новых мутаций являлись причиной изменения аминокислот и были обнаружены у единичных пациентов или целых семей. Фактически, это обусловило некоторую неопределенности в их интерпретации. Поэтому поиск оптимального способа исследования влияния CLCN7- мутаций на функции белков является насущной необходимостью, чтобы различать редкие полиморфизмы и мутации и правильно составлять корелляцию генотип-фенотип. Кроме того, как упоминалось выше, был выявлен синонимичный вариант в 12 экзоне CLCN7, вызывающий частичную аберрацию сплайсинга с пропуском 12 экзона внутри рамки. Полученные сведения привели к появлению гипотезы о том, что некоторые синонимичные, но не явно патогенные изменения способны модулировать фенотип мутаций CLCN7. Для подтверждения этой теории может понадобиться анализ клинических и генетических данных крупной когорты пациентов с CLCN7-зависимым остеопетрозом.
SNX10
Ген SNX10 (сортирующий нексин 10) кодирует белок, относящийся к семейству SNX – цитоплазматических и мембраносвязанных белков, характеризующихся наличием фосфоинозитид-связывающего домена PX. Как правило, SNX-белки участвуют в сортировке белков и мембранном транспорте, устанавливая связи белок-белок и белок-липид. В частности, SNX10 взаимодействует с V-АТФазой и регулирует ее внутриклеточный транспорт; таким образом, АРО с дефицитом SNX10 возникает в результате нарушения транспорта V-АТФазы к фестончатому краю и последующей дисфункции остеокластов. Также в очень редких случаях SNX10 может предположительно играть роль в транспорте и секреции матриксной металлпротеазы 9 при деградации внешнеклеточного матрикса.
Мутации гена SNX10 служат причиной около 5 % всех случаев АРО, в том числе т.н. Вестерботтенского остеопетроза (по названию провинции в Швеции с высокой частотой возникновения данного заболевания). В частности установлено, что вариант SNX10 c.212+1G>T, вызывающий активацию криптического сайта сплайсинга в 4-ом интроне и аберрантного сплайсинга, является общей мутацией для данной когорты пациентов, с необыкновенно высокой частотой носителей (1:93) в общей популяции Вестерботтена. Примечательно, что генеалогические исследования и гаплоидный анализ свидетельствуют в пользу наследования данной мутации от общих предшественников в начале 19-го века.
На клеточном уровне, несмотря на полученные ранее противоречивые данные по поводу наличия или нехватки остеокластов при АРО с дефицитом SNX-10, Статтин с соавт. говорят не о дефекте дифференциации остеокластов в МКПК пациентов, а о нарушении формирования фестончатого края. В то же время, остается под вопросом предположение, что у человека инактивация SNX10 приводит к остеопетрозному рахиту (о чем свидетельствуют исследования на мышиной модели), поскольку лишь у нескольких пациентов присутствует данный специфический фенотип. В очень редких случаях индуцированные плюрипотентные стволовые клетки (ИПСК) перепрограммируются из кожных фибробластов (у пациента с мутацией c.212+1G>T). Данные ИПСК могут способствовать изучению не раскрытых до конца особенностей SNX10-зависимого остеопетроза.
OSTM1
Ген OSTM1 (остеопетроз-ассоциированный трансмембранный белок 1) кодирует трансмембранный белок I типа, локализованный, в основном, на эндосомах и лизосомах. Он обладает высоко гликолизированным N-концом, который стабилизирует CLCN7 и защищает его от деградации лизосом, а также играет важную роль в обмене 2Cl− /1H+. Трансмембранный домен задействован в ионном обмене и CLCN7-зависимом транспорте к лизосомам. Предполагается также, что OSTM1 действует в качестве убиквитин-лигазы E3 для гетеротримерного G-белка Gαi3 и усиливает канонический сигнальный путь WNT с помощью модуляции взаимодействя β-катенин/ Lef1.
Недавно были выявлены дополнительные цитозольные партнеры OSTM1 по связыванию, что позволило предположить, что OSTM1 может служить в качестве адапторной молекулы внутри цитозольного поддерживающего мультипротеинового комплекса.
Мутации гена OSTM1 являются причиной примерно 5 % всех случаев АРО и всегда вызывают крайне тяжелый фенотип с быстро прогрессирующей первичной нейродгенерацией. Практически все выявленные мутации данного гена являются дефектами усечения. В связи с этим, при усечении OSTM1 в скрытой форме наблюдается ингибирование формирования остеокластов in vitro путем снижения числа осей BLIMP1-NFATc1, что, возможно, приводит к другим патогенным механизмам в случае АРО с дефицитом OSTM1. Более того,благодаря специально разработанному количественному ПЦР-анализу, две различных гомозиготных микроделеции, перекрывающие ~ 110 и ~ 10 кб соответственно и поражающие N-терминальный участок гена OSTM1, обнаружены у 5 тяжело больных пациентов из двух неродственных семей арабского и индийского происхождения. Анализ последовательностей релевантного геномного участка выявил AluSx-опосредованную рекомбинацию и нереккурентную перестройку, сопровождающуюся негомологичным соединением концов, в соответствии с лежащим в основе молекулярным механизмом.
Крайне редко у пациентов описывается остеопетроз с ранней нейродегенерацией и аккумуляцией железа в специфических областях головного мозга, что является крайне необычным случаем. Полное экзомное секвенирование выявило новые c.783+5G>T мутации гена OSTM1, вызывающие пропуск 4-го экзона, а также мутации сдвига рамки c.446dup в гомозиготном состоянии гена MANEAL. Данный ген кодирует эндо-альфа-подобный белок маннозидазы, который предположительно локализован в аппарате Гольджи и потенциально включен в метаболизм гликопротина; и действительно, в моче и цереброспинальной жидкости пациентов было обнаружено повышенное содержание маннозных молекул тетрасахариды. До сих пор неясно, как это соотносится с аккумуляцией железа в головном мозге. Влияние мутации гена MANEAL на фенотип, ассоциированный с OSTM1, в целом, требует дальнейшего изучения.
PLEKHM1
Ген PLEKHM1 (плекстрин гомологичное доменсодержащее семейство М – с RUN доменом 1) кодирует цитосолический белок, задействованный в эндосомальных транспортных путях ввиду взаимодейтсвия с малыми ГТФазами RAB7 и ARL8. Помимо этого, PLEKHM1 участвует в слиянии аутофагосом и лизосом, необходимом для клиренса различных белковых агрегатов. В соответствии с этим, разрушение специфичных PLEKHM1-доменов или потеря PLEKHM1 нарушает секрецию и доставку везикул, а также формирование фестончатого края, что ухудшает резорбтивную функцию остеокластов.
PLEKHM1 – это крупный белок, который содержит различные функциональные домены: домен RUN, где локализованы мутации (NM_014798.2:c.296+1G>A), изначально определенные у двух сиблингов с АРО; 2 плекстрин-гомологичных (PH) домена, отделенных друг от друга LC3-взаимодействующей областью (LIR); рубикон-гомологичный (RH) домен и «цинковый палец» С1 в карбоксильном конце. Две различные предположительно доминантные мутации гена PLEKHM1 выявлены у двух неродственных пациентов: мутация c.2140C>T:p.Arg714Cys, неявно ассоциированная с остеопетрозом, обнаружена во втором PH-домене; и недавно определенная мутация c.3051_3052delCA, расположенная в RH-домене и предположительно ликвидирующая мотив цинкового пальца. RH-домен необходим для взаимодействия PLEKHM1 с RAB7; в подтверждение этого, исследования сверхэкспрессии в клетках HEK293T показали снижение взаимодействия мутировавших белков с RAB7, приводящее к аномальной внутриклеточной локализации и повышению уровня аутофагии. Эти сведения внесли важный вклад в понимание функции PLEKHM1 для биологии костных клеток, несмотря на то, что некоторые аспекты и, в частности, взаимосвязь с аутофагией, остаются спорными и нуждаются в дальнейшем изучении.
CAII
Ген САII кодирует цитоплазмический фермент, катализирующий получение H2CO3 с помощью CO2 и H2O; затем, H2CO3 диссоциирует на HCO3 и ионы H+. Полученный H+ вытесняется V-АТФазой, в то время как HCO3 поглощается HCO3 − and H+ - анионным обменником, расположенным в базолатеральной мембране, которая предотвращает алкалинизацию цитоплазмы и обеспечивает ионы Cl−, необходимые для CLCN7/OSTM1 2Cl− /H+ антипортера.
Помимо того, ген CAII в больших количествах экспрессируется в почках и головном мозге. Фактически, пациенты с дефицитом CAII демонстрируют остеопетроз, почечный тубулярный ацидоз (ПТА) и церебральную кальцификацию, и данная триада нарушений сама по себе формирует диагноз. Примечательно то, что проксимальный ПТА недавно был описан также у пациента с АДО II, который является носителем распространенной при АДО II p.Gly215Arg мутации и у которого не было других мутаций, ассоциированных с ПТА; в данной ситуации точный патогенный механизм до сих пор остается неясным.
В процессе стандартизации условий для генетического тестирования выявлено около 30 различных мутаций CAII: миссенс-мутации, стоп-мутации и мутации сайта сплайсинга (среди последних - NM_000067.2:c.232+1G>A или т.н. арабская мутация, из-за широкой распространенности среди пациентов арабского происхождения). Большинство пациентов с мутацией CAII имеют арабское происхождение, однако встречаются также европейцы, испанцы, афро-амерканцы, азиаты и, реже, китайцы Хань.
FERMT3
Ген FERMT3 (член семейства фермитина 3) кодирует киндлин-3, член семейства киндлина, который включает 3 различных белка фокального контакта, задействованных в активации интегрина. Это процесс, необходимый для клеточных адгезии (слипания), распластывания и миграции, организации внутриклеточного матрикса, а также выживания, пролиферации и дифференциации клеток. Киндлин-3 – внутриклеточный белок, связанный с актиновым цитоскелетом. Он взаимодействует с множеством классов интегринов и способствует их адгезивной функции и сигнализации изнутри наружу, которая необходима костной ткани для резорбтивной функции остеокластов. Таким образом, дефицит киндлина-3 вызывает тяжелую морфологическую альтерацию остеокластов и нарушает их способность прилипать к поверхности костей. Интересен тот факт, что мутации гена FERMT3 ответственны за редкое аутосомно-рецессивное заболевание под названием дефицит адгезии лейкоцитов (LAD-III), некоторые пациенты с LAD-III также страдают от тяжелой формы остеопетроза (10 из 23 опубликованных случая). Эти мутации в основной массе являются высоко разрушительными: мутации усечения, дефекты сплайсинга, сдвиг рамки; из всей когорты выявлены только 2 миссенс-мутации. К сожалению, поскольку число описанных в литературе случаев ограничено, в настоящее время невозможно провести корреляцию генотип/фенотип.
RANKL
Ген RANKL (лиганд рецептора-активатора ядерного фактора каппа-B) предназначен для фундаментальных остеокластогенных цитокинов, которые путем связывания с его рецептором RANK запускают активацию нисходящего сигнального каскада, вызывающего дифференциацию и активацию остеокластов.
Недавно был найден дополнительный рецептор молекулы RANKL, богатый лейцином повтор-содержащий G-белок-связанный рецептор 4 (LGR4), который в результате связывания лигандов активирует сигнальный путь GSK3, тем самым подавляя экспрессию NFATc1 в ходе остеокластогенеза. Кроме того, были открыты ранее неизвестные и неожиданные функции RANKL в костной ткани, играющие важную роль в остеогенетической дифференциации мезенхимальных стволовых клеток, возможно, с помощью аутокринной петли.
Таким образом, полученные сведения представляют большой интерес для изучения АРО с дефицитом RANKL - редкой формы остеопетроза, которая составляет всего 2 % всех случаев. Для лучшего понимания упомянутых молекулярных механизмов и их значения в патофизиологии кости необходимы дальнейшие исследования.
По имеющимся данным, новых мутаций гена RANKL и соответственно случаев заболевания, помимо уже известных, до сих пор не было выявлено.
RANK
Как уже упоминалось выше, ген RANK (рецептор-активатор ядерного фактора каппа-B) является функциональным рецептором для RANKL.Связывание с лигандом вызывает тримеризацию рецептора и рекрутинг различных адапторных молекул, а также активацию различных сигнальных путей, таких как c-Jun N-терминальной киназы (JNK)/белок-активатор-1 (AP-1), ядерный фактор каппа-B (NF-kB) и ядерный фактор активированных Т-клеток с1 (NF-kB), Src и p38/MITF, Src and ERK, в результате чего происходит дифференциация, активация и выживание остеокластов. Мутации гена RANK ответственны приблизительно за 5% случаев АРО. В отличие от АРО с дефицитом RANKL, при котором костные дефекты не лечатся ТГСК, при АРО с дефицитом RANK ТГСК является эффективной, как показали недавно полученные данные.
SLC29A3
Дисостеосклероз – это редкая форма остеопетроза, которая развивается в младенчестве и проявляется типичными скелетными признаками: (расширение концов трубчатых костей и платиспондилия), а также различными поражениями кожи. Клиническое течение и прогнозы, как правило, довольно благоприятные. Имеющиеся литературные данные касательно генетики диостеосклероза довольно ограничены. Ген SLC29A3 (глюкозный траспортер-3 семейства 29), который при дисостеосклерозе поражен, кодирует нуклеотидный транспортер лизосом, в больших количествах вырабатываемый в клетках миелоидной линии. Описываемые мутации (NM_018344:c. 607T>C:p.Ser203Pro и c.1157G>A:p.Arg386Gln у одного пациента и c.1346C>G:p.Thr449Arg у второго в гомозиготном состоянии, а также c.303_320dup:p.102_107dup в гомозиготном состоянии) могут оказывать влияние на функцию и дифференциацию остеокластов, на что указывает сниженное число остеокластов после дифференциации in vitro МКПК пациентов, а также образцов биопсии кости. Мутации SLC29A3 также ассоциируются с гистиоцитозом с массивной лимфаденопатией – группой заболеваний, не затрагивающих или лишь слегка затрагивающих скелет. Таким образом, необходимы дальнейшие исследования для лучшего понимания роли данного гена в костной ткани, и особенно – в патофизиологии в целом.
CTSK
Пикнодизостоз - это редкая аутосомно-рецессивная скелетная дисплазия (примерная частота случаев - 1:1,7 млн. человек), которую включают в дифференциальную диагностику остеопетроза из-за повышенной плотности костной ткани в трубчатых костях. Другие типичные для пикнодизостоза клинические признаки включают низкорослость, которую с переменным успехом можно лечить гормонами роста, незакрытые родничок и швы черепа, переломы, тупой нижнечелюстной угол и акроостеолиз дистальной фаланги. При данном заболевании поражен ген CTSK, кодирующий катепсин К – цистеиновую пептидазу суперсемейства папаин, используемую остеокластами для деградации костного матрикса и обладающую уникальной способностью расщеплять молекулы коллагена на маленькие фрагменты. Кроме того, недавно было обнаружено, что катепсин К расщепляет и активирует матриксную металлопротеиназу 9 in vitro, что может указывать на наличие протеаз-сигнальной сети, вероятно релевантной для ряда физиопатологических нарушений. Также последние исследования показали, что катепсин-K играет роль в регуляции моделирования кости путем деградации периостина - белка внутриклеточного матрикса кортикального слоя, необходимого для формирования надкостницы при участии сигнального пути Wnt-β-катенина.
До сих пор в литературе было описано около 60 различных мутаций CTSK у пациентов из различных географических регионов. Наиболее частыми вариантами являются миссенс-мутации, также выявлены сдвиг рамки, нонсенс-мутации и дефекты сплайсинга. Мутации возникают, в основном, в зрелых CTSK-белках, с «горячими точками» в 5 и 6 экзонах.
Кроме того, около 6% мутаций обнаружено в пререгионе и 25 % - в прорегионе – небольших N-терминальных доменах, необходимых, соответственно, для правильной локализации и укладки белков и внутриклеточного транспорта. Прорегион также важен для поддержания ферментов в неактивном состоянии, при низком уровне pH он расщеплен. При этом корреляция генотип-фенотип до сих пор остается неизученной до конца.
Гены, вовлеченные в недавно открытые синдромные формы остеопетроза
TRAF6
Наряду с различными адапторными молекулами, необходимыми для связывания RANKL/ RANK, TRAF6 (TNF-рецептор-ассоциированный-фактор-6) представляется наиболее важной. Действие TRAF6 также снижает продукцию рецепторов Т- и B-клеток, что приводит к активации NF-kB. Несколько лет назад было определено, что деактивация гена TRAF6 у мышей вызывает тяжелый остеопетроз, и, крайне редко, схожее действие наблюдалось и у человека. Фактически, гомозиготная делеция 2064 Kb в 11-ой хромосоме, перекрывающая 5 регион в генах TRAF6, RAG1 и RAG2 (белки RAG необходимы для рекомбинации рецепторов B и T клеток, а также для их выживания и дифференциации) обнаружена у 2 сиблингов с остеопетрозом и тяжелым комбинированным иммунодефицитом (ТКИД) с помощью хромосомного микроматричного анализа. Точные границы делеции определены не были; что же касается TRAF6, его геномная делеция включала регион выше 1 экзона и часть некодирующей последовательности экзона 1. Данные участки с большой вероятностью являются регуляторными. Фактически, на белковом уровне их делеция полностью отменяет продукцию TRAF6. Результирующий фенотип является специфичным, поскольку остеопетроз у пациентов с данной группой мутаций был выражен исключительно в костях таза и ног. Поскольку в обоих случаях летальный исход наступил в очень раннем возрасте в связи с тяжелым иммунодефицитом и в настоящее время подобные случаи неизвестны, проследить развитие скелетного заболевания с данной генетической базой затруднительно.
LRRK1
Остеосклеротичная метафизарная дисплазия является формой остеопетроза, поражающей метафиз длинных костей, концевые пластинки позвонков, оконечности ребер, а также края плоских костей, в то время как череп остается непораженным. Описано только 5 случаев данного заболевания; в одном из них недавно была определена гомозиготная делеция семи нуклеотидов в последнем экзоне гена LRRK1 (лейцин-богатая повторная киназа 1) (NM_024652:c.5938_5944delGAGTGGT:p.Glu1980Alafs*66), предположительно вызвавшая сдвиг рамки и преждевременную терминацию, с потерей седьмой триптофан-аспартической кислоты (WD) 40-го домена. Домен WD-40, также как остальные функциональные домены белка LRRK1, выступает посредником во взаимодействии белок-белок. В частности, LRRK1 предположительно взаимодействует с компонентом сигнального пути c-Src для достижения цитоскелетной перестройки и формирования фестончатого края и подосомы. Таким образом, у мышей остеокласты с дефицитом Lrrk1 являются крупными и плоскими, поскольку они неспособны должным образом осуществить реорганизацию цитоскелета и резорбцию кости. Ген LRRK1, в целом, представляет высокий интерес для костной биологии и дальнейшие исследования должны определить его физиопатологическую роль.
MITF
Микрофтальмия-ассоциированный транскрипционный фактор (MITF) представляет собой транскрипционный фактор с основным доменом типа спираль-петля-спираль (лейциновая застежка). Он формирует гомо- и гетеродимеры, регулирующие генную экспрессию в различных тканях. По этой причине его мутация может вызвать широкий спектр фенотипов. В костной ткани MITF предположительно действует совместно с сигнальным путем RANKL/RANK, уменьшая продукцию NFATc1 для усиления NFATc1-зависимого остеокластогенного сигнала. И действительно, для мышей с Mitf mi/mi характерен остеопетроз в связи с дефектом остеокластогенеза на ранних стадиях.
Сложные гетерозиготные мутации гена MITF совсем недавно были обнаружены у двух неродственных пациентов с т.н. COMMAD-синдромом (колобома, остеопетроз, микрофтальмия, макроцефалия, альбинизм и глухота). Выявленные мутации (NM_198159.2:c.952_ 954delAGA:p.Arg318del and c.921G>C:p.Lys307Asn у пробанда I; c.952A>G:p.Arg318Gly и c.938- 1G>A:p.Leu312fs у пробанда II) повлияли не на димеризацию MITF, а скорее на его миграцию внутри ядра и ДНК-связывающие свойства. Данное открытие указывает на ряд фенотипов, связанных с MITF; фактически, в отличие от вариантов с рецессивными мутациями, доминантные ассоциированы с синдромом Ваандербурга типа 2A и синдромом Тица, для которых характерны глухота и пигментный дефицит. В целом, эти данные подтверждают ключевую роль MITF в процессах развития, а также дифференциации и выживания клеток.
Сигнальный путь NF-kB
Сигнальный путь NF-kB включает ряд молекул (в основном, киназы и транскрипционные факторы), которые играют ключевую роль в экспрессии регуляторных генов во многих органах и физиопатологических процессах. В костной ткани на это указывает тот факт, что гипоморфные мутации гена NEMO, кодирующего компонент IkB-киназного комплекса, необходимого для ингибирования IkB-α и последующей ядерной транслокации продуцируемого гетеродимера p65/p50, ответственны за Х-сцепленный остеопетроз с эктодермальной дисплазией и иммунодефицитом. Данные мутации в основном локализованы в белковом домене «цинковый палец» и приводят к остеопетрозу по причине альтерации сигнального пути RANKL/RANK.
Недавно было выявлено, что белок p65 (Rela) сам по себе связан с фенотипом, характеризующимся высокой костной массой. Фактически, в случае с внезапным летальным исходом новорожденного пациента, патологическое увеличение костной массы, выявленное в результате аутопсии и обоснованное повышенной функцией остеобластов, как оказалось, было ассоциировано с de novo миссенс-мутацией гена RELA (NM_021975.3:c.1534_ 1535delinsAG:p.Asp512Ser). Данная мутация, по наблюдениям, нарушает сигнализацию NF-kB в фиробластах пациентов, и возможно, может привести к нарушениям различных жизненно важных функций.
CSF1R
Наряду с RANKL, M-CSF является ключевой остеокластогенной молекулой. Это можно явно наблюдать при остеопетрозе с нехваткой остеокластов у мышей с дефицитом данного цитокина (мышиная модель op/op). У мышей дефицит M-CSF-рецептора (CSF1R) приводит к сходному остеопетрозному фенотипу; кроме того, обе модели демонстрируют дефекты врожденного иммунитета, фертильности и неврологической функции.
Примечательно то, что доминантные мутации гена CSF1R вызывают взрослую форму энцефаломиопатии, в то время как совсем недавно подозревалось, что именно рецессивная мутация данного гена ответственна за летальный, тяжелый фенотип у 2 сиблингов с генерализированным остеопетрозом и мальформацией мозга в тяжелой форме.
Вкратце, секвенирование экзома близкородственных родителей выявило гетерозиготную мутацию усечения (NM_001288705.1:c.1620C>T:p.Tyr540*) в гене CSF1R, который предположительно активировал белок с нехваткой внутриклеточного домена, необходимого для лиганд-зависимой димеризации и аутофосфореляции. В условиях отсутствия образцов ДНК, гомозиготность мутации CSF1R у пациентов не могла быть продемонстрирована; по этой причине, полученные результаты нельзя считать полностью обоснованными. Тем не менее, на их основании возможно проанализировать гены других пациентов со сходным фенотипом, чтобы попытаться определить дополнительные мутации в качестве подтверждения.
C16orf57
Несколько лет назад был описан случай итальянского пациента, у которого был обнаружен остеопетроз, ассоциированный с пойкилодермией и нейтропенией (ПН). ПН-синдром - это наследственный генодерматоз, который характеризуется ранней пойкилодермией, дистрофией ногтей, ладонно-подошвенным гиперкератозом и устойчивой нейтропенией, приводящей к рекуррентным инфекциям. В подобных случаях могут присутствовать скелетные дефекты, такие, как остеопения, краниофациальный дисморфизм, переломы, постнатальная задержка созревания и роста скелета. Однако случай, описанный Мильяччо с соавт. оказался нетипичным. ПН-синдром был обусловлен мутациями гена C16orf57, кодирующего фосфодиэстеразу, ответственную за модификацию и стабилизацию U6 небольшой ядерной РНК (USB1), которая является необходимым элементом структуры сплайсомы.
Позднее у пациента с остопетрозом и ПН была обнаружена гомозиготная стоп-мутация в гене C16orf57 (ENSG00000103005:c.232C>T:p.Arg78*), предположительно вызвавшая серьезные нарушения укладки процессированного белка. Выявленный патогенетический механизм может быть неполным или аберрантным сплайсингом специфических классов генов, как можно предполагать учитывая нейтропению. Тем не менее, для подтверждения данной гипотезы необходимы дальнейшие исследования скелетных клеток.
Современные методы терапии
В настоящее время единственным утвержденным методом лечения АРО является трансплантация гемопоэтических стволовых клеток (ТГСК). Остеокласты, фактически, являются клетками гемопоэтического происхождения, поэтому ТГСК способствует восстановлению костной резорбции с помощью донорских клеток. Данная статья не предусматривает подробного описания метода ТГСК: режимов кондиционирования, источников ГСК, типов донорства, графика и других, без сомнения важных, аспектов. Подробную информацию о них можно найти в недавно вышедших работах мировых экспертов. Здесь будут лишь вкратце приведены некоторые концепции.
Во-первых, молекулярные исследования вносят важный вклад в определение того, в каких случаях следует проводить ТГСК. Фактически, пациенты с мутациями RANKL или OSTM1 не могут быть кандидатами на данную процедуру: в первой группе дефекты не являются клеточно-автономными и поэтому не могут быть скорректированы с помощью ТГСК, а во второй у пациентов присутствовали серьезные неврологические нарушения несовместимые с жизнью. Для подобных случаев в настоящее время терапии, к сожаллению, не существует.
Кроме того, тяжесть клинической картины диктует метод лечения: легкие формы, ассоциированные с PLEKHM1, SLC29A3 или CTSK или доминантными мутациями CLCN7 не являются основанием для рискованных инвазивных процедур, проводимых при ТГСК. В таких случаях применяется только симптоматическое лечение в случае осложнений (переломов, дефектов зубов, отоларингологических проблем). С другой стороны, при АРО с дефицитом CLCN7 рекомендована тщательная неврологическая оценка с целью принятия решения о релевантности ТГСК. Установленная прогрессивная дегенерация препятствует проведению трансплантации. К сожалению, в настоящий момент, в данной субгруппе не установлена специфичная корелляция генотип-фенотип, позволяющая выявить пациентов, подходящих для ТГСК на основании лишь молекулярных данных.
Наконец, что касается недавно диагностированных форм заболевания, пока недостаточно данных для определения специфической стратегии лечения в дополнение к основной.
Выводы
Последние данные подтверждают генетическую неоднородность остеопетроза, осложняющую молекулярную диагностику. Фактически, в патогенез остеопетроза включены более 10 генов, включая самые крупные. Среди наиболее часто поражаемых определено большое количество различных мутаций: некоторые из них присутствуют у многих пациентов различного происхождения, другие - в единичных случаях, также определены новые мутации. По этим причинам, секвенирование следующего поколения (NGS), включая полное экзомное секвенирование (WES) и таргетированное секвенирование с использованием аналитических панелей, прочно вошло в клиническую практику в качестве стандартной процедуры диагностики остеопетроза. Комплексный метод имеет явные преимущества в скорости и более приближен к точной молекулярной диагностики, необходимой для прогнозирования и лечения, по сравнению с классическим секвенированием по Сенгеру генов-«кандидатов».
Кроме того, с помощью WES в единичных случаях у пациентов с нетипичными фенотипами удалось определить мутации в новых генах, которые, как уже известно играют роль в гомеостазе костной ткани: TRAF6, LRRK1, MITF, CSF1R и RELA. Генетические данные при этом указывают на возможную, но еще до конца не определенную роль C16orf57 в костях. В некоторых случаях, базисный патогенный механизм все еще нуждается в дальнейшем изучении, несмотря на то, что по литературным данным можно достаточно обоснованно судить об отрицательном воздействии выявленной мутации. С другой стороны, 2 класса мутаций, а именно глубокие интронные или синонимичные изменения, демонстрируют возможные ограничения WES. На самом деле, даже несмотря на то, что многие некодирующие участки являются целью WES, стандартный процесс по отбору вариантов рассматривает только интронные изменения, близкие к каноничным сайтам сплайсинга; в особенности, тенденцию оставаться незамеченными имеют варианты в конце прочтения.
Поэтому, если мутация локализована в данных регионах, она может быть пропущена. Фактически, данная работа ясно демонстрирует у 2 неродственных семей патогенный эффект глубоких интронных однонуклеотидных изменений, обнаруженных в гене TCIRG1, который сначала был пропущен WES. Схожим образом синонимичные изменения часто отфильтровываются в ходе стандартного технологического процесса при WES. На самом деле, они часто игнорируются также при стандартном секвенировании по Сенгеру, поскольку предположительно являются «тихими». Испытуемая группа продемонстрировала ( и это совпадает с результатами других исследований), что данное предположение в некоторых случаях может быть в корне неверным.
В заключении можно отметить, что совершено значительное достижение в изучении генетической базы остеопетроза. Тем не менее, около 10 % случаев все еще нуждаются в молекулярной классификации и у данных пациентов могут быть обнаружен мутации новых, ранее неизвестных генов либо неявные мутации в известных. Для того, чтобы восполнить пробелы, необходимо найти стратегию по интеграции NGS-технологий и улучшению способности интерпретировать геномные данные. В комплексе это позволит определить клинически релевантные варианты среди десятков тысяч в каждом отдельном экзоне.

Остались вопросы?
Напишите нам и мы свяжемся с вами в течение ближайшего времени и ответим на все интересующие вопросы